Snowflake ML 中的 CUDA-X 库¶
使用 Snowflake Container Runtime 的 CUDA-X 集成,以通过 GPUs 无缝扩展数据转换和 ML,而无需更改代码。Snowflake 已将 NVIDIA 的 cuML 和 cuDF 库集成到运行时环境中。 通过这种集成,您可以将 scikit-learn、umap-learn 或 hdbscan 等库与 GPUs 结合使用。您无需学习新框架或处理复杂的依赖项。
您可以运行主题建模、基因组学和模式识别等复杂处理,而不会影响数据大小或算法复杂性。缩短处理时间使您有机会进一步迭代模型。
通过与 CUDA-X 库集成,在 Snowflake ML Container Runtime 中会实现大型数据集的 GPU 加速处理。 处理速度可能比单独使用 Container Runtime 快几个数量级。
适用于数据科学的 NVIDIA CUDA-X 库¶
开源库(例如 cuML 和 cuDF)利用 GPUs 实现更高效和可扩展的数据工作流程。您可以使用这些库来处理具有数十亿行和数百万维度的数据。有关这些库的更多信息,请参阅 NVIDIA CUDA-X 数据科学 (https://developer.nvidia.com/topics/ai/data-science/cuda-x-data-science-libraries)。
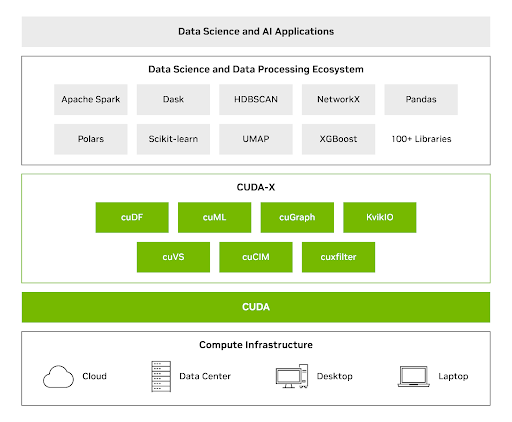
CUDA-X DS 库将 GPUs 的强大功能与常用的 Python 库相结合来进行数据分析、机器学习和图形分析,无需团队重写代码即可大幅提升速度。借助 CUDA-X DS,您可以使用 GPU 速度提升,通过单个 GPU 处理高达 TB 大小的数据集。
NVIDIA cuML 可通过 CPU 工作流程提供以下性能改进:
scikit-learn 提升到 50 倍
UMAP 提升到 60 倍
HDBSCAN 提升到 175 倍
用例¶
Snowflake ML Container Runtime 中 CUDA-X 库的集成使用 GPUs 与 Scikit-learn 和 pandas 来实现以下用例:
大规模主题建模¶
对大型且特征丰富的数据集进行主题建模需要:
使用嵌入模型
大规模应用降维
使用聚类和可视化提取准确且相关的主题
GPU 并行性可以帮助您更高效地完成上述工作流程。通过使用 cuML 加速处理,您可以将数以百万计的产品评论从原始文本转换为定义明确的主题群集,这些主题群集可以从 CPU 上的数小时减少到 GPU 上的几分钟,而无需修改现有 Python 代码。这突出了 UMAP 和 HDBSCAN 库的无缝插入式加速。
有关通过 Snowflake 上的 GPUs 执行主题建模的更多信息,请参阅 https://quickstarts.snowflake.com/guide/accelerate-topic-modeling-with-gpus-in-snowflake-ml/#0 (https://quickstarts.snowflake.com/guide/accelerate-topic-modeling-with-gpus-in-snowflake-ml/#0)
计算基因组学工作流程¶
使用 Snowflake 的 CUDA-X 集成可显著加速生物序列的处理。您可以将 DNA 序列转换为特征向量,用于可扩展的分类任务,例如预测基因家族。
在使用 cuDF 和 cuML 在 GPUs 上直接执行 Pandas 和 scikit-learn 代码,可加速数据加载、预处理和集成模型训练。此 GPU 无需更改代码即可加速现有工作流程,使研究人员能够优先考虑生物学见解和模型设计,而不是低级别的 GPU 编程。
在 Snowflake 中开发¶
使用 CUDA-X 库在 Snowflake ML Container Runtime 中开发和部署 GPU 加速型机器学习模型。本章节提供了将这些工具集成到 Python 工作流程中的分步指南。
要开始使用,请执行以下操作:
在 Snowflake 笔记本或 ML 作业中定义 Python 脚本
为笔记本或 ML 作业选择相应的 GPU 运行时和 GPU 计算池。
完成上述步骤后,运行以下代码在您的环境中配置 CUDA-X 加速器。
现在,您可以通过 GPUs 直接运行 Pandas 操作,或者拟合 scikit-learn、umap 或 hdbscan 模型(请注意,无需更改代码即可通过 GPUs 运行)。此示例演示如何在大型数据集上使用 hdbscan:
应用用例:大规模主题建模¶
计算效率对于大规模文本分析和主题建模至关重要。GPUs 使用并行处理将处理时间从几小时缩短到几分钟。本章节演示了通过使用 CUDA-X 的 GPU 加速,如何在包含 200,000 条美容产品评论的数据集上加速 ML 模型。
您可以使用 CUDA-X 执行以下操作:
将原始文本转换为机器学习的数字表示形式(嵌入)。
加速降维
要利用 CUDA 库,请在代码开头添加 %load_ext cuml.accel。这会将处理时间从几小时缩短到几分钟。
以下示例代码使用 SentenceTransformer 类来创建嵌入。
以下示例代码使用 HDBSCAN 以减少高维数据。它会保留群集主题。
应用用例:运行复杂的基因组学工作流程¶
基因家族组织(包括旁系同源物和直向同源物)对于理解基因进化、功能和保守的生物过程至关重要。
借助 CUDA-X 库,您可以创建分类模型,以根据 DNA 序列预测基因家族。该模型可以加速基因组注释,识别新的基因功能,并提供对进化途径的见解。
数据集 (https://raw.githubusercontent.com/nageshsinghc4/DNA-Sequence-Machine-learning/master/human_data.txt) 具有一系列纯文本核苷酸序列及其相应的基因家族类标签。这些类对应七个不同的人类基因家族。
以下代码使用 Hugging Face 中的 核苷酸转换器 将 DNA 序列转换为向量。该转换器对序列进行标记化和批处理,将每个基因序列转换为 1280 个特征向量。
您可以使用以下代码来评估两个集成分类模型:
随机森林分类器
XGBoost 分类器